




Running CrystalPredictor 2.4.3.2 with torsion groups
Torsion groups
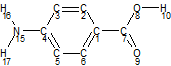
For some molecules, independent degrees of freedom can be so far separated that they have no inter-dependence. Consider, for example, aminobenzoic acid. The amine group and the acid group are a long way from one another, and highly unlikely to affect one another. If you wanted the amine group to have H_N_C_C vary from -40 to +40 in 10 degree steps (for a -45 to +45 search space), and the acid group to have O_C_C_C vary from -40 to +40 in 10 degree steps and the H_O_C_C angle vary from -80 to +260 in 20 degree steps (for a -90 to +270 search space), this would be a three dimensional grid of 9*9*18 (1458) LAM points. However, if you could treat the two areas of the molecule separately, you would have one grid of 9 LAM points (for the amine group) and another of 162 LAM points (9*18). The computational saving of this grid should be apparent!
Versions available on cposs
There are two different versions of CrystalPredictor that work with torsion groups, 2.4.3.2_torgrp and 2.4.3.2_torgrpcon. Louise thinks you can use the latter in all cases, but has not tested this. The difference is in the ability of the latter to also constrain other degrees of freedom (for example methyl groups) which would also save a lot of time in LAM generation.
You can run any version with one flexible and one rigid molecule, but you currently cannot run one molecule with torsion groups and one flexible but without torsion groups. If you find yourself in this situation, see if you can add a torsion group to the second molecule. It will be more computationally expensive, but will enable you to run!
Input files
Input files are the same as for the regular version of CrystalPredictor, and are described in this document. The differences are set out below.
input.in
This file is exactly the same as for a normal flexible search. Just ensure that the degrees of freedom are in the same order as in your molecule.in file.
potential.in
Again, exactly the same as any other search.
molecule.in
This section describes the molecule.in file you would use for your search. You need to generate the databases of LAM points for the torsion groups separately. The instructions for generating each torsion group is the same as for the normal flexible searches, so your starting molecule.in files would be different and not refer to the torsion groups.
Intramolecular energy/gradients/hessian/charges for: | The heading. This title doesn't even need to match that in input.in. |
Generated at level of theory: | This is where you specify the level of theory for the calculation of the LAM grid. |
Across 2 dimensional grid: | This is where you tell CrystalPredictor that you have two different torsion groups and which angles are included in each. |
Constraints: 4 |
If you are adding any additional constraints (such as methyl rotations) you need to use version 2.4.3.2_torgrpcon |
From starting Z-matrix: | Then follows the Z-matrix, which is exactly the same as for a normal flexible molecule. |
cc2 1.3945 | Then follows the starting points for the variables, WITHOUT a blank line between the Z-matrix definition and the variables. The independant degrees of freedom for Group 1 and then Group 2 come next, followed by any Constraints. (This example is slightly messy as dih49 is the independent degree of freedom for Group 1, but would also be the last degree of freedom if it was dependent.) |
###TOR GROUP 1######################################### | Then come the LAM points. You will need to put these in manually. Follow the instructions for generating LAM points for the two different torsion groups. Then append the LAM points from the first group and the LAM points from the second group. The separator lines are shown here, or you can consult the example in ~cposs/CrystalPredictor/example_inputs/ Don't worry that ENTRY 00001 appears twice - the number is not used for anything! |
LAM generation
The databases of LAM points are generated for the two torsion groups separately (in separate directories), with the independent degrees of freedom of the other torsion group being treated as Constraints. The molecule.in file is NOT as described above, but as described in the instructions for running without Torsion groups. In each case, you will have the independent degrees of freedom being analyzed in this group, the independent degrees of freedom being analyzed in the other group(s), and then any constraints. Append the LAM databases as described in the previous section or as seen in the example file on cposs.
Header section for the molecule.in file for the first torsion group: |
Intramolecular energy/gradients/hessian/charges for: |
Header section for the molecule.in file for the second torsion group: | Intramolecular energy/gradients/hessian/charges for: |
© UCL Chemistry Department 2022. This page was last updated on 17 August, 2022. If you have any problems with this page please email the WebMaster