




Packing Coefficients
For some of the systems we study, it will make more sense to calculate packing coefficients than density. The packing coefficient is the amount of the unit cell that is occupied by the molecule, and for hard spheres is around 74%. The advantage of calculating the packing coefficient is that it is possible to compare systems with different molecular weights to see whether there is potential for solvent to be included.
There are two methods of calculating packing coefficients available to the group. One is the CCDC software provided by Aurora, and the other is using Platon.
The CCDC software is available on both Xenon and Blackadder in /home/cposs/UTILITIES/packingcoefficients/ and is run using the following command
/home/cposs/UTILITIES/packingcoefficients/bin/packingcoefficients structure.res 0.1
where 0.1 is the grid spacing in Angstroms. This can be adjusted if necessary.
Platon is only currently available on Xenon. To run it, type
platon structure.res
This will allow you to run commands, such as "CALC VOID" to calculate the packing coefficient and void space. To exit, just press Enter without typing a command. The grid spacing is always 0.2 angstroms. It is apparently possible to change according to the manual, which can be found at http://www.cryst.chem.uu.nl/platon/
Method of calculation
Both the CCDC software and Platon use the same method for calculating the packing coefficient. The positions of the centres of each atom are know, along with the species of atom, and a model of the molecule can be generated.
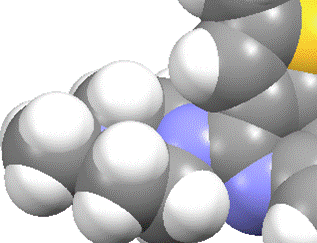
A grid is generated, with spacing as defined in the command for the CCDC software or with spacing of 0.2 Angstroms in the case of Platon.
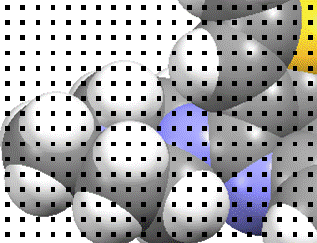
The points that are within and without the molecular spheres are differentiated. The percentage of points inside the molecule is the packing coefficient.
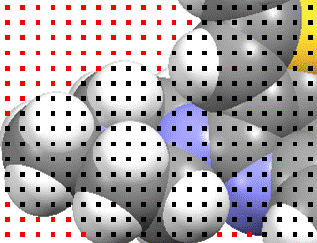
Platon includes extra steps, where any point that is more than a certain distance from the edges of the spheres is differentiated further (blue points), and those that are within this probe radius distance of such a point included too (green).

The blue and green points would include the limits of any solvent molecule that might fit. The method is described in full in Acta Cryst. A 46 194-201 (1990).
Difference between CCDC software and Platon
The main difference between the two methods is in the atomic radii used. The software the CCDC distributes is using on atomic radii calculated by Gavezzotti in J. Am. Chem. Soc. 105 5220-5225 (1983). These are one for each type of atom - all carbon atoms have the same radius, and all hydrogens have the same radius. Platon is more sophistocated, and uses a different atomic radius for atoms of different type, as tabulated in Bondi’s work J. Phys Chem. 68 441-451 (1964).
The CCDC software is therefore significantly less accurate than Platon, but is much faster because it identifies the atoms from the SFAC line of the .res file rather than analysing each for the bound atoms in order to identify it. One of the minor problems with the CCDC software is that some of the historical files on the CPOSS data store have incorrect SFAC lines.
The version of Platon on Xenon has problems with some packing types.
Further comments, including plots of the correlation between the different methods of calculating the packing coefficient and density, are found in PackingCoefficientsComments.doc
© UCL Chemistry Department 2022. This page was last updated on 17 August, 2022. If you have any problems with this page please email the WebMaster