




Notes on how to perform extended searches
The purpose of this document is to explain how to construct all necessary input files to perform a search for lattice energy minima using CrystalPredictor and refine them with more accurate intermolecular potentials using multipoles. CrystalPredictor can perform searches assuming rigid or flexible molecules. If molecular flexibility is important then the final refinement can be initially done with DMAFLEXquick which does not require any wavefunction calculation and finally with CrystalOptimizer which is more accurate but takes significantly longer. The idea here is to reduce the number of hypothetical minima kept as the accuracy of the model is increased. Hence expensive calculations are only performed on structures whose energy is low enough to be of significance. The number of structures to keep when we move from one step to the other depends on the system and cannot be decided a priori. We need to examine how much re-ranking is caused when a change in the computational model is introduced. For some systems (e.g. non polar molecules) there is no re-ranking at all when potential derived charges are replaced by distributed multipoles. On the other hand, for systems that are mainly bound by electrostatic interactions (hydrogen bonding and more importantly molecular salts) the re-ranking when charges are replaced by multipoles is significant and hence hundreds or thousands of structures need to be kept when we move from one step to the other. The formal mathematical background on the procedure outlined in this document can be found in Mol.Phys. 105, 273-291, 2007. For rigid bodies there is no need to construct intramolecular potentials which simplifies the problem to a large extent. If the molecule can be considered rigid please proceed to section 1.3 that describes how to set up searches for stable packing arrangements.
Note: CrystalPredictor is limited to 60 atoms. We think that this is 60 atoms per molecule, and that you may have more than 1 molecule in the system with a total number of atoms exceeding 60. We do not know whether a molecule of greater than 60 atoms, split into two fragments each of fewer than 60 atoms, is acceptable - if you know either way, please email Louise to update this note.
1 Considering flexibility
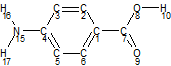
CrystalPredictor1.9 can be used to perform searches on rigid or flexible molecules. In the case of flexible molecules, the molecule is split into a set of rigid fragments. Consider aminobenzoic acid:

The molecule can be split into rigid fragments. Each rigid fragment contains at least one flexible bond, and the atoms at either end of each flexible bond are contained in two fragments. In the case of an amine group, it is desirable to consider the two hydrogen atoms separately, as the group can be planar or pyramidal. This gives the set of rigid fragments shown below:

In the case of aminobenzoic acid, the flexibility of the amine group on the left is highly unlikely to affect the flexibility of the ethanoic acid group on the right. Hence the two parts of the molecule can be treated independently when assessing the intramolecular flexibility.
2 Folder structure and required files for CrystalPredictor1.x
It is important that the files are in the correct directory structure for CrystalPredictor1.6
- Your search directory
- molecules.in
- space_groups.in
- potential.in
- intramolecular potentials (for flexible molecules only)
- charges files (for searches using variable charges)
- input (folder)
- input.in
- unix_executable (folder)
- runCrystPred_1.9.sh
- runAnalyse_1.9.sh
2.1 molecules.in
molecules.in file for a rigid molecule
molecules.in file for a flexible molecule
alternative file ending for a flexible molecule with torsion groups
This file defines the molecular geometries and the structure of the asymmetric unit. Even in the case of the rigid molecule, you need to include the torsion definition section, and leave it blank.
For most problems the number of distinct molecular types is 1, unless the study involves co-crystals (then it is 2), salts (it is 2), monohydrates (it is 2), monohydrate co-crystal (it is 3) etc. Most other entries are self explanatory apart probably the field determining the molecular geometry. The first column is the number of the atom. The second column is the potential type (it should correspond to the entries in the file potential.in). The third column is the symmetry equivalence of the atom, for example in benzene all 6 carbon atoms would have a Tsym label of 1, and all 6 hydrogen atoms would have a Tsym label of 2, because of the six-fold axis. If the molecule is flexible then this column is (usually) identical to the first. The fourth column is the atomic weight. The fifth column is the atomic charge, which should be computed for the global conformational minumum (or the local conformational minimum for the conformational region under study). This column is completely ignored if you use variable conformation charges as discussed in section 2.6. The last three columns are the Cartesian coordinates (in Angstrom) of the atoms. In the case of flexible molecules, CrystPredictor will use these coordinates to set up the geometry of the rigid fragments. A good choice is to give the coordinates of the global conformational minimum, or the local conformational minimum for the conformational region under study.
rigid fragments and torsion groups are explained in the section on intramolecular potentials.
The next field determines the number of rigid fragments, which is 5 for aminobenzoic acid. The following field gives the atoms that belong to each fragment. In the example input file for a flexible molecule the first fragment comprises the aromatic ring. You can use any order for the fragments, although the performance of the minimization is improved if the first fragment (called mother fragment) is chosen to lie centrally in the molecule.

Finally, you need to specify the number of torsion groups. If the molecule is complex and there are flexible parts in large distances you can assume that the intramolecular energy can be approximated as a sum of energy penalties to deform non-interacting flexible parts. This can dramatically reduce the number of quantum mechanical calculations to construct the intramolecular potentials. For example, in the case of aminobenzoic acid we could assume that the carboxylic group and amine functional group geometry are energetically decoupled. The ending of the molecules.in file would then be as given here.

If the torsion angles are split in more than one group, then CrystalPredictor will either require constant charges or variable charges that depend only on the torsion angles of the first group. The current version of CrystalPredictor allows up to 4 torsions in each torsional group.
2.2 space_groups.in
This file can be copied from /home/cposs/CrystalPredictor1.9/ex-input/spacegroups.in on cposs. The top of the file has a list of all the available spacegroups (commented out), and you can delete any you don't wish to cosider from the bottom.
2.3 potential.in
CrystalPredictor allows the use of exp-6 and 12-6 potentials. The above example corresponds to the latest Williams potential which is of exp-6 type. The atom types should match the atom types give in molecules.in. Note that if cross terms are not given they are computed with standard mixing rules.
The potential.in file containing the FIT potential can be copied from /home/cposs/CrystalPredictor1.9/ex-input/potentials.in on cposs. If you want to use the Will01 potential, copy williams_pot.in from that folder and call it potential.in
2.4 intramolecular potentials file(s)
These are only required if your molecule is flexible, and you are considering flexibility in the search. You first need to consider the flexibility within the molecule, and whether you are able to split the molecule into separate parts with different torsion groups.
Consider aminobenzoic acid:
The molecule can be split into rigid fragments. Each rigid fragment contains at least one flexible bond, and the atoms at either end of each flexible bond are contained in two fragments. In the case of an amine group, it is desirable to consider the two hydrogen atoms separately, as the group can be planar or pyramidal. This gives the set of rigid fragments shown below:
In the case of aminobenzoic acid, the flexibility of the amine group on the left is highly unlikely to affect the flexibility of the ethanoic acid group on the right. Hence the two parts of the molecule can be treated independently when assessing the intramolecular flexibility.
For each torsion group, the other part of the molecule which we are treating as unrelated can be substituted by a hydrogen (or methyl or whatever is appropriate). Each torsion group can only have up to 4 flexible angles.
LAMs should be generated for the molecule or each torsion group as described in the CrystalPredictor2.2 notes. We don't think that non-uniform grids are allowed with CrystalPredictor1.x. Then the lam2cp16 executable can be run to convert the format (this can be found in the /home/cposs/CrystalPredictor1.9/grid_utilities folder on cposs or in the /home/cposs/CrystalPredictor/for1.8grid folder on cposs). This will give you the intramolecular potential file, called intra.out. You will need to rename this to intra_x_y.pot, where x is the molecule number and y is the torsion group number within your system. Hence the aminobenzoic acid example would require files called intra_1_1.pot and intra_1_2.pot.
example of intra_1_1.pot for aminobenzoic acid, without separating ito torsion groups.
This file has 4 degrees of freedom, with 5 possible values for the first, 5 possible values for the second and so on. Then each lines gives an index number, the value of each flexible degree of freedom, the energy for that combination, and the gradient with respect to each degree of freedom in turn.
2.4.1 Extending the grid
Occasionally, a CrystalPredictor1.9 search will reach the limits of the flexible degree of freedom and the minimization will terminate. In this case, it is useful to put an artificially high "wall" on the end of the intra_1_1.pot file. In the example shown here, the user might, for example, want to extend the first degree of freedom to -60 and +60 (it currently has values of -40, -20, 0, 20 and 40). In this case, for every combination of the second, third and fourth degrees of freedom, you would need to add -60 and +60 with energies of 100 and gradients of zero (indicating that it is a perfect maximum and this point). There are utilities to do this for 3 dimensional (ext_grid-3) and 4 dimensional grids (ext_grid) in /home/cposs/CrystalPredictor1.9/grid_utilities/ on cposs or /home/cposs/CrystalPredictor/for1.8grid on xenon. You can do it manually for 1 or 2 dimensional grids!
Examples of an original (3 dimensional) grid and its extended counterpart (new rows are in yellow and changes in cyan) are available.
2.5 charges file(s)
If you want to run CrystalPredictor with variable charges, the charges should be calculated for each point on the intramolecular potential grid. However, the gradients of these charges are not used from the file as the user provides but are calculated incorrectly within CrystalPredictor. This leads to very odd results, with resulting crystal structures being in bands of energy. At the moment, it is not recommended that they are used.
Because it is not recommended that they are used, we currently do not extract them from the LAMs to make charge_1.pot files. The information is available in the lam_intra file, and the format required is given in this example file for aminobenzoic acid (which contains 17 atoms, hence 17 charges at each combination of flexible degrees of freedom). Note that there is only one index in the filename. The reason is that, if you have multiple torsion groups, the atomic charges of the whole molecule are assumed to depend on only the values of the torsion angles that belong to the first torsion group.
2.6 input.in
It is very important in CrystalPredictor1.x that input.in is put in a subdirectory called input. If it is not there, the code will not find it and it will fail.
Most of the fields are self explanatory. The output and input directory are given relevant to the location of the executable “CrystPred”. The section “SIMULATION” provides data to the minimizer. In this example we choose 15 A cut off for the vdW interactions. However, we initiate a cubic spline at 12 A for the interaction to drop smoothly to zero over the distance range 12-15 A to ensure a continuous objective function. The real space electrostatic energy is computed up to a distance of 15 A; the rest of the electrostatic energy is computed on reciprocal space. CrystalPredictor adjusts the number of reciprocal vectors based on the cut off for the real part and the value of the Ewald summation accuracy. The choices shown in this example “input.in” file are fine for most systems. The next parameter defines the pressure (typically 0 Pa for most studies). The next keyword determines whether the hydrogen bondlengths X-H should be adjusted to standard neutron values. If you use quantum mechanically computed molecular coordinates in “molecules.in” you do not need to standardize. The next keyword determines whether the X-H bondlengths should be foreshortened for the lattice energy minimization. For most studies it is recommended to use Williams potential and hence this keyword should be set to “yes”. The final keyword of the “SIMULATION” section is to define whether to use “constant” charges or “variable”, conformation-dependent charges. We have found that there is a problem with variable charges, as the interpolation is not calculated correctly. CrystalPredictor calculates the gradients of the charges internally (we're not sure how or why), and seems to get them wrong, leading to severe steps in the energies. You should use constant charges unless you know this has been fixed. If the molecule is rigid then the keyword should be set to “constant”.
The final, “SEARCH” section of the input determines the behaviour of the putative structure generator. The first four entries are the minimum and maximum lattice angles and lattice lengths. For most systems these values are sufficient unless there are multiple molecules in the asymmetric unit in which case you may wish to increase the maximum cell length to increase efficiency. The next parameter is the minimum density; if a putative structure once generated has density lower than the specified value it is discarded without being minimized. Similarity, a putative structure is discarded if its initial intermolecular energy or intramolecular energy penalty are greater than the specified thresholds (in this case 50 kJ/mol and 40 kJ/mol). The putative structure is also discarded if it corresponds to a cell with particularly acute or obtuse angles; in this example input file all cells with a deformation parameter less than 0.15 are discarded (this is a good choice for most problems). The next number defines the number of minimizations to perform. If this is set to small number then the global optimization may be incomplete. Tell-tale signs for an incomplete search maybe the presence of several low-energy structures that have been encountered only once. In such cases CrystalPredictor allows to continue the search starting from the point the calculations ended as describe in section 1.3.6. The last number is the range of polymorphism. In this example all minimized structures that are less stable than the global minimum (at any given time) by at least 15 kJ/mol are discarded. You may want to increase this value if you expect large re-ranking if multipoles are used, as is for example the case with salts or other strongly hydrogen-bonded systems.
3 Running CrystalPredictor
3.1 unix_executable folder
You should copy the entire unix_executable directory from /home/cposs/CrystalPredictor1.9 on cposs, as a subdirectory of your search folder. You can change the jobnames and the number of processors on which CrystalPredictor will run in the runscripts.
The number of allocated processes is specified in the line “#$ -pe openmpi 20”. Note that CrystalPredictor needs at least 4 processes to run.
3.2 Submitting the job
To submit the job enter qsub runCrystPred_1.9.sh
3.3 Checking the progress
The submission script is configured so that all input/operators are done in the frontend. This is ok because there is only minimal amount of information being written and no scratch space used for the calculations. Hence, to check the progress you do not need to login to any node. One way to check the progress is to copy the tool see from /home/cposs/CrystalPredictor1.9/ into the directory where the results are stored and type ./see x where x is number of molecules in the asymmetric unit.
3.4 Restart capabilities (file “restart.chk”)
In the unfortunate situation that the number of minimizations chosen in the file “input.in” is not sufficient, CrystalPredictor can be restarted without repeating the minimizations already done. This is done by reading the file “restart.chk” if it is present. This file is always created once CrystalPredictor terminates. To restart you need to rename some files as follows:
- mv crystals.out crystals1.out (assuming this is the first restart)
- mv crystals_stable.out crystals_stable1.out
- mv global_statistics.out global_statistics1.out
to ensure that results from the incomplete run are not overwritten.
4 Output files and post processing
CrystalPredictor creates several files, the most important of which are as follows:
- crystals.out, which contains a record for each minimized structure regardless of the final energy
- crystals_stable.out, which contains only the structures within the range of polymorphism. This file will be post-processed to construct initial guesses for DMACRYS, DMAFLEXquick and CrystalOptimizer refinement depending on whether the system is flexible or not.
- log.out, which contains all messages from CrystalPredictor. You do not need to examine this file unless spurious results and unexpected behaviour is observed.
To post-process the results, from your unix_executable directory enter qsub runAnalyse_1.9.sh Analyse inspects each structure in crystals_stable.out to see whether it is unique, and for each unique structure it writes one or two files to a unique_pool folder created in the search directory. If a structure is found more than once, then the corresponding cluster (assume the cluster number x in ascending energy order) is represented with the two files minimum_x_1.pdb and minimum_x_2.pdb that are equivalent, although they may correspond to different crystallographic setting. If the cluster only contains one structure then only minimum_x_1.pdb is created. The pdb files can be visualized with Mercury. Analyse also writes the structures in spf (PLATON) format that is handy for further-processing. Usually it is sufficient to process only one file from each cluster (i.e. “minimum_x_1.spf”). The second file is to provide an alternative starting point if the refinement fails, which does not happen very frequently.
One final step before refinement is to convert the spf files into res files that DMACRYS/DMAFLEXquick/CrystalOtpimizer understand. This is done by copying /home/cposs/CrystalPredictor1.9/post-processing/Imperial_Rename_BA.sh into the directory above unique_pool and submitting it to the queue. For each cluster found by CrystalPredictor ImperialRename creates one folder named “x-1” that contains the required res file named as “x.fres”. These res files need to be refined with DMACRYS/DMAFLEXquick/CrystalOptimizer.
There is an old document which may still be of some use (if the codes still exist on cposs!)
5 Example output
The following are probably no longer available, but I will look at rerunning this example and putting all the files somewhere central.
An example of crystal structure prediction for aminobenzoic acid with one and two molecules in the asymmetric unit can be found in:
- /home/pk/CrystalPredictor/Aminobenzoic_acid/Zpr_1, a Z’=1 search with atomic charges held constant and computed for the MP2/6-31G(d,p) charge density of the HF/6-31G(d,p) conformational minimum. Only the 13 most probable space groups were considered. The search involved 50k minimizations and took around 5 hours on blackadder using 13 processes.
- /home/pk/CrystalPredictor/Aminobenzoic_acid/Zpr_2_both_hands, a Z’=2 search with the two molecules in the asymmetric unit having opposite chirality. The intramolecular energy file (intra_2_1.pot) for the molecule with opposite chirality can be constructed by inverting the sign of the torsion and gradients values of the intra_1_1.pot file. Moreover, all x,y,z coordinates of the second molecule in molecules.in are constructed by inverting the sign of the coordinates of the first molecule.
- home/pk/CrystalPredictor/Aminobenzoic_acid/Zpr_2_same_hands, a Z’=2 search with both molecules in the asymmetric unit having the same chirality.
© UCL Chemistry Department 2022. This page was last updated on 17 August, 2022. If you have any problems with this page please email the WebMaster